
Context
| MASH compute approximate distances between genomic sequences. First, it utilizes the MinHash technique to reduce genomes to compressed sketch representations. Then, using only the sketches, which can be thousands of times smaller, similarities between sequences can be rapidly estimated. | 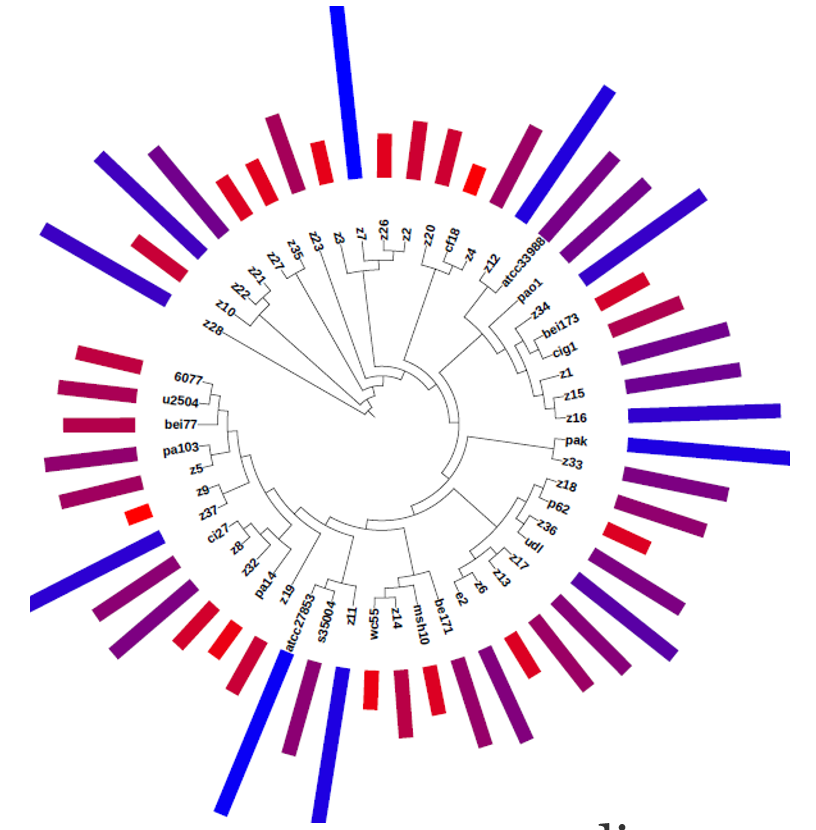 |
Code link
Reference
-
Ondov, B.D., Treangen, T.J., Melsted, P. et al. Mash: fast genome and metagenome distance estimation using MinHash. Genome Biol 17, 132 (2016). https://doi.org/10.1186/s13059-016-0997-x